One of the continuing scandals in the physical sciences is that it remains impossible to predict the structure of even the simplest crystalline solids from a knowledge of their composition.I previously posted about this "scandal".
Molecular crystals are particularly challenging because they often form polymorphs, i.e. several distinct competing crystal structures, that have energies differing on the scale of 1 kJ/mol [0.2 kcal/mol ~ 10 meV]. Calculating the absolute and relative energies of different crystal structures to this level of accuracy is a formidable challenge, going far beyond what is realistic with most electronic structure methods, such as those based on Density Functional Theory (DFT).
This problem is also of great interest to the pharmaceutical industry because the "wrong" polymorph of a specific drug can not only be ineffective but poisonous.
Aside: Polymorphism is also relevant to superconducting organic charge transfer salts. For example, for a specific anion X the a compound (BEDT-TTF)2X multiple crystal structures are possible, denoted by greek letters alpha, beta, kappa, ... , with quite different ground states: superconductor, Mott insulator, metal, ...
There are two key components to the computational challenge of predicting the correct crystal structure:
1. having an accurate energy landscape functionPreviously, it was thought that 2. was the main problem and one could use semi-empirical "force fields" [potential energy functions]. However, with new search algorithms and increases in computational power it is now appreciated that 1. is the main bottleneck to progress.
2. searching the complex energy landscape to find all the low-lying local energy minima
Sally Price has a nice tutorial review of the general problem here.
Every few years there is a crystal structure prediction competition where several teams compete in a "blind test". A report of the most recent competition is here.
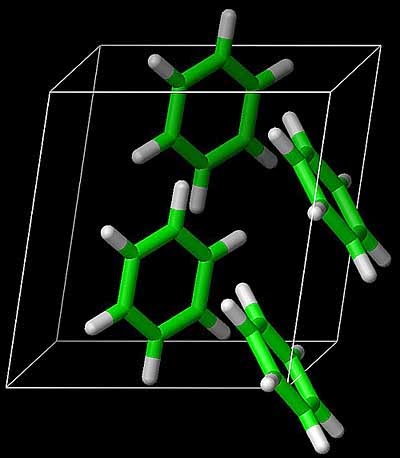
Garnet described recent work in his group [soon to be published in Science] using state-of-the-art computational chemistry methods to calculate the sublimation energy of benzene [the hydrogen atom of organic molecular crystals].
It is essential to utilise two advances in computational chemistry from the last decade:
1. local correlation theories that use fragment expansions
2. explicit r12 correlation.
They obtain a ground state energy with an estimated uncertainty of about 1 kJ/mol.
This agrees with the measured sublimation energy, although there are various subtle corrections that have to be made in the comparison, including the zero-point energy.
Update. August 11, 2014.
The paper has now appeared in Science with a commentary by Sally Price.